Perelman School of Medicine | University of Pennsylvania

| NGSC - FAQs |
Next-Generation Sequencing Core Perelman School of Medicine | University of Pennsylvania |
|
Test your antibody and sample prep before making a library. We recommend that the enrichment ratio \( {{C^+/I^+}\over{C^-/I^-}} \ge 10 \). Where \( C^+ \) is ChIP at positive control, \( I^+ \) is input at positive control, \( C^- \) is ChIP at negative control, and \( I^- \) is input at negative control.
To do this, you need at least two primer pairs, a positive control (+) and a negative control (-) which you measure on the ChIP and input samples using Q RT-PCR. See the figure to the right.
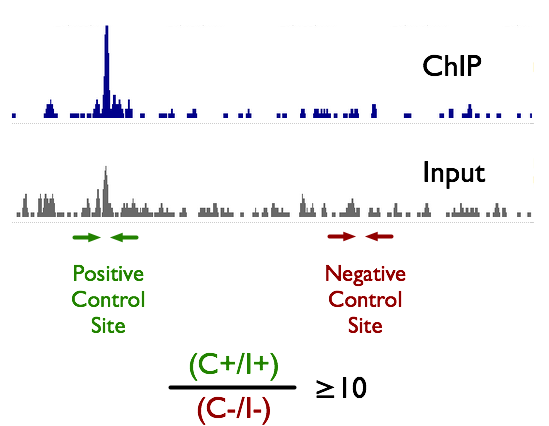
We strongly recommend that you sequence an \textcolor{red}{input} library for each condition. Note that in the figure, the input track has a strong peak at the same place as the ChIP peak. The strength of inputs peaks or bias is dependent on the state of the cells as well as chromatin preparation conditions.
Like most Illumina library preps, ChIP-Seq libraries usually require a number of rounds of PCR. PCR creates copies of DNA fragments which are statistically dependant on each other. During data analysis it is important to reduce dependant fragments to a single fragment when possible. This can reduce the read count dramatically for some libraries. However duplicate fragments can also be produced in ways that are statistically independant and so can be left in during the analysis, if the analysis is able to identify which is which.
Usually DNA is fragmented by sonication for ChIP-Seq libraries which produces largely random breakage. So in regions with low coverage, duplicated reads are more likely to come from PCR copies. As the coverage gets deeper, the chance that a duplicated read is from an independent fragment increases, though only if the library has sufficient complexity. In enriched areas, i.e., peaks, it is quite possible to see duplicate reads that are independant.
Since removing duplicated reads can dramatically reduce the effective depth of sequencing it is important to assess the level of duplication and take steps to reduce it if necessary and if possible. However, the amount of material available, the size of the target's footprint in the genome, and the effectiveness of the anitbody may limit what is possible. The table below covers most of the common scenarios.
| Redundancy in Peaks | Redunancy in Background | Summary | Suggestions |
| no peaks | high | IP is not working, and little background material is available | IP conditions may be too stringent or antibody does not work at all. |
| no peaks | low | IP is not working, but plenty of background material is available | IP conditions may be not stringent enough or antibody does not work at all. |
| low | high | not possible | |
| low | low | plenty of material available prior to PCR. | You can work with data as is, or consider adjusting IP conditions to be more stringent if enrichment could be stronger. |
| high | high | Little material prior to PCR | If effective read counts are workable, can be left as is. Otherwise consider using more cells or pooling multiple IP before library prep |
| high | low | Enrichment should be very strong so redundancy in peaks is due to crowding or pigeon-hole principle. | Data should be usable as is, but if you are measuring differential binding, you may want to take advantage of redundany reads. Also suggests that you may be able to use less chromatin in the IP. |