Perelman School of Medicine | University of Pennsylvania

| NGSC - FAQs |
Next-Generation Sequencing Core Perelman School of Medicine | University of Pennsylvania |
|
FASTQ is the file format the NGSC uses to store read sequence and quality data. This Wikipedia article is a good source for the details of the file format. Beyond the basic format, it is handy to note that the files we produce include the defline information that allows you to uiquely identify every read. Also the article describes the evolving read quality metrics that Illumina generates. This is relevant for our older data.
We name the primary FASTQ files by the run, lane, barcode, and for paired-end sequencing read. This allows us to generate unique file names that are safe for any filesystem.
RUN_s_LANE_BARCODE.fastq.gz FGC1103_s_7_CTTGTA.fastq.gzRUN_s_LANE_END_BARCODE.fastq.gzFGC1099_s_1_1_ACAGTG.fastq.gz read 1FGC1099_s_1_2_ACAGTG.fastq.gz read 2Clearly this naming system does not identify the sample the data is for. To
make this connection, see the AAA-StudyInfo.xls or AAA-StudyInfo.txt file
in the root of your investigation. It lists all of the samples that have been
submitted to the investigation. If the samples have been sequenced there is a
separate line for each run and lane. (See the RULA_Run and RULA_Lane
columns.) The barcode is in RULA_Barcode. Runs or lanes that failed will have
an F in the RULA_Status column. The RULA_Geometry column indicates the
machine type, mode, serial number, and sequencing geometry. See the following
table of sequencer serial numbers and models.
Occasionally the wrong machine is listed here. So it is best to check for the machine's serial number in the FASTQ deflines.
| Serial Number | Machine Type | Active |
| M00590 | MiSeq | yes |
| NS500618 | NextSeq 500 | yes |
| D00712 | HiSeq 2500 | yes |
| K00315 | HiSeq 4000 | yes |
| --- | --- | --- |
| SN628 | HiSeq 2000 | no |
| SN423 | HiSeq 2500 | no |
| SN431 | HiSeq 2500 | no |
| SN965 | HiSeq 2000 | no |
| SN969 | HiSeq 2000 | no |
| SN1160 | HiSeq 2000 | no |
Data that was not associated with a barcode is placed in files with Undetermined in the name. In a typical experiment this would be either the PhIX library reads or reads where the barcode was misread and count be reliably associated with a known barcode. In less typical experiments, e.g., where the NGSC does not know the barcodes this file may contain all of the interesting data.
The FASTQ files located under basic/FASTQ have not been trimmed in any
way - poor quality reads are included as well as adapter sequence. Each read
should be the same length. Some of our downstream analysis pipelines will do
trimming, but that data is located elsewhere and may take a few days to appear.
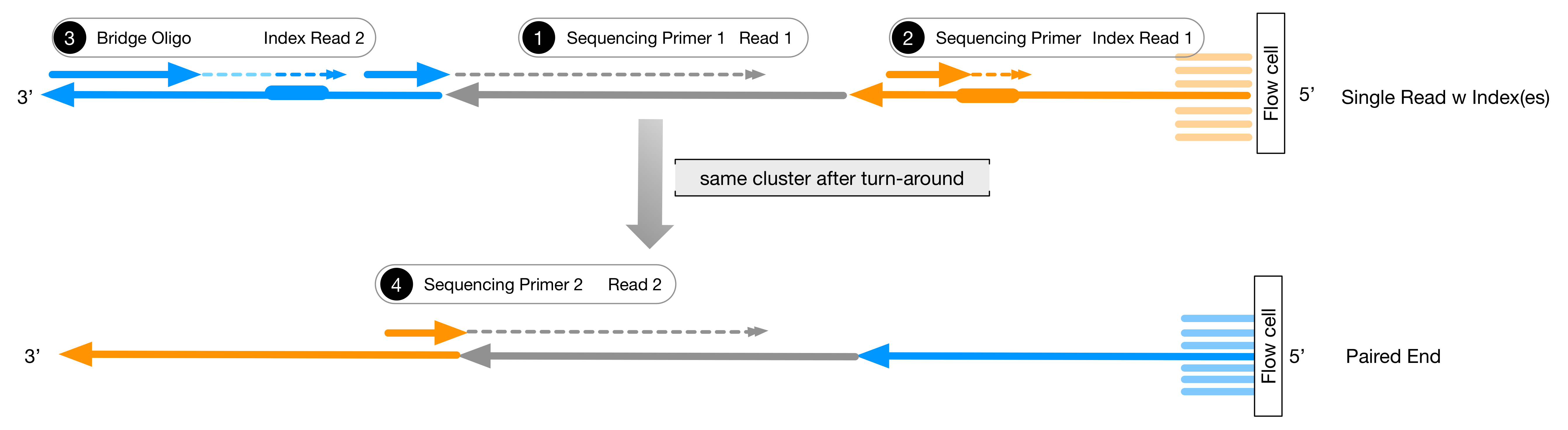
Illumina sequencers sequence libraries as shown in the figure above. There are up to four reads from each fragment. The reads are indicated by the dashed lines with the double arrow-head.
Gray read is read 1 of the insert.
Orange read is the first index, aka barcode read.
Blue read, if using dual indexing, is the second index read
The second Gray read, in paired-end sequencing, is a second read from the other end of the insert.
Each of these reads has a fixed length which is defined at the start of the run. The index
reads are always set to read just the barcodes which have a fixed length. The insert will
usually vary in length. Since the length of the insert read is always the same length, a
100bp (for example) read will include the first 100bp of the insert. In the figure, the
insert is longer than the read, but this it not always true. If the insert is shorter than
100bp, the read will contain the leading part of the 3' adapter sequence. If the total length
of the insert and the 3' adapter is less than 100bp, then the sequening polymerase will hit
the flow-cell wall and stall. At this point there will be a series of 6 to 8 As followed by
nonsense bases. During the 'polyA' stretch the base quality drops to zero. In paired-end
sequencing, the second read will follow a similar pattern but will sequence into the 5'
adapter. If the insert is shorter than twice the read length, then the reads will overlap at
their ends (on opposite strands.)
Note that since the barcodes are read with dedicated reads, they are not normally included in
the insert read. Illumina's bcl2fastq program uses only the dedicated index reads to
determine which sample the read belongs to. The index read data is placed in the defline of
the read(s).
The barcode information we maintain for each sample is the barcode sequence as it will be sequenced on the majority of Illumina sequencers. Thus, following the scheme in the figure, if the 3’ barcode is visible in the read, it appear in the same sense as the file name and our database annotation. However, the 5’ barcode will appear in reverse compliment sense in insert read 2 (if visible) since on all machines (except the NextSeq) index read 2 and insert read 2 are done in opposite directions. For trimming it is usually easiest to look for the inner end of the adapters then trim everything from there on out. That way you do not have to supply a different sequence for each barcode.
When demultiplexing data for a lane, we generally allow 1 mismatch between the
sequenced barcode and the target barcode. For example a read with a barcode
ATCGTA would be placed in the file with barcode ATCGTG. Barcodes that
differ by more than 1 mismatch are placed in the Undetermined file. If, due
to a library construction, sample annotation, or pooling error, an incorrect
set of barcodes is given to the demultiplexing process, then there may be a
large number of reads in the Undetermined file. A quick command line check of
the barcode frequency in this file can identify the presence of one or more
unexpected barcodes.
In order to save disk space and make copying faster we compress FASTQ files
using gzip. A technical detail of the compression is that is done on chunks
of 10 million reads and the chunks are concatenated together. This is perfectly
ok in most cases, but can cause two problems.
Some browsers only decompress the first chunk, so your decompressed file only contains 10 million reads.
The remedy is to switch to a different browser.
The checksum file we also generate for each FASTQ file (with .sha256sum at
the end) is made from the chunky file. If we or you decompress and recompress
the file, then the checksum of the new file will not match the chunky one.
There is no easy remedy for this. For this reason we are changing our piplines to generate single chunk files
The NextSeq 500 is different from the other Illumina sequencers in two important ways that impact the FASTQ files it generates.
The NextSeq 500 has 4 lanes. Each lane gets the same sample or pool, but they are imaged by different cameras. Therefore, the data is tagged with lane numbers 1 to 4. However, the data in each file is for the same sample and represents distinct set of fragments for the sample. We generally keep these files separate, but not always.
The NextSeq 500 sequences the second read of a dual-indexed library in the reverse direction from the other sequencers. We reverse complement the second barcode in the file name, but not in the FASTQ deflines.
So for example, a barcode pair TAAGGCGA and TAGATCGC would be sequenced
as TAAGGCGA and GCGATCTA. The defline for a read would contain
TAAGGCGA-GCGATCTA but we would rename the FASTQ file to TAAGGCGATAGATCGC.